Genetic diseases of the central nervous system

In our genetics clinic, we see a large variety of patients with rare diseases, with a focus on neurodevelopmental and neurodegenerative disorders. We study the mutations in our patients in combination with their phenotypes in order to understand gene function in humans. We also use reprogramming of patients’ cells and differentiate induced pluripotent stem cells into neural precursor cells, neurons and cerebral organoids to study gene function and the mechanisms of disease. We put a particular emphasis on understanding the molecular mechanisms underlying variability of clinical phenotypes. Mouse models and analysis in patient cohorts complete our methodological repertoire. With all these attempts, we aim to develop experimental therapies for patients with rare disorders. Some of the diseases we study are:
Early processes in Huntington’s Disease
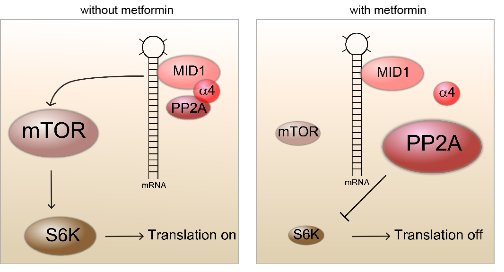
Huntington’s Disease (HD) is a late-onset and devastating neurodegenerative disorder that is very hard to detect in the early stages. However, once the disease has reached the symptomatic phase, neurodegeneration is already far advanced and therapy is likely to be too late for older patients. Using mouse models of HD, we have found aberrations in the cortical network at a very early stage before disease onset, which were associated with subtle behavioural abnormalities. We had found that the synthesis of disease-causing protein in Huntington’s Disease is driven by a protein complex that contains the mTOR kinase (mammalian target of rapamycin). Metformin inhibits formation of this complex and, as we could show, substantially reduces the production of disease-causing protein in an animal model of Huntington’s Disease. We are currently investigating whether early treatment with metformin can improve later disease progression in the mouse and have put together a clinical trial for patients before disease onset.
Epigenetic enhancement of non-canonical gene expression in Birk-Barel Syndrome
Mutations in the imprinted gene KCNK9 cause Birk-Barel mental retardation syndrome. Using a mouse model of the disease, we characterised the allele-specific expression of Kcnk9 in various regions of the brain and correlated this with brain function and behaviour. We observed partial expression of the paternal allele in several brain regions and intermediate phenotypes in animals with a maternal-only knockout of Kcnk9, leading us to hypothesise that stimulating paternal gene expression could rescue the phenotype. Using specific histone deacetylase inhibitors, we induced upregulation of the paternal Kcnk9 allele in primary cortical neurons and in the brains of Kcnk9 KOmat mice. This fully rescued the cognitive deficits, suggesting that this may be a promising experimental therapy for patients with Birk-Barel mental retardation syndrome.
Dynamic X-chromosomal reactivation enhances female brain resilience
Sexual dimorphism is well-documented in neurodevelopmental disorders, but the underlying molecular mechanisms are not well understood. One of the most important differences between male and female mammals is the sex chromosomes. In order to allow dosage compensation between the sexes, large parts of one X chromosome are randomly inactivated in females. We use induced pluripotent stem cells, neural precursor cells, neurons and brain organoids as models to study how X chromosomal gene expression and inactivation are regulated during neurodevelopment.
In collaboration with the ReALity community and with Claudia Keller-Valsecchi (IMB), Felicia Basilicata (IMB), Joan Barau (IMB) and Peter Baumann (IMB/JGU), we also plan to study X chromosomal gene re-activation in the developing immune system and work to understand the molecular mechanisms underlying X chromosomal re-activation.