Barau Lab
Research Publications Group Members BiographyTransposable elements & genome stability
Our lab is broadly interested in how the genetic conflict with transposable elements affects the regulation and stability of the mammalian genome during development and evolution.
Transposable elements
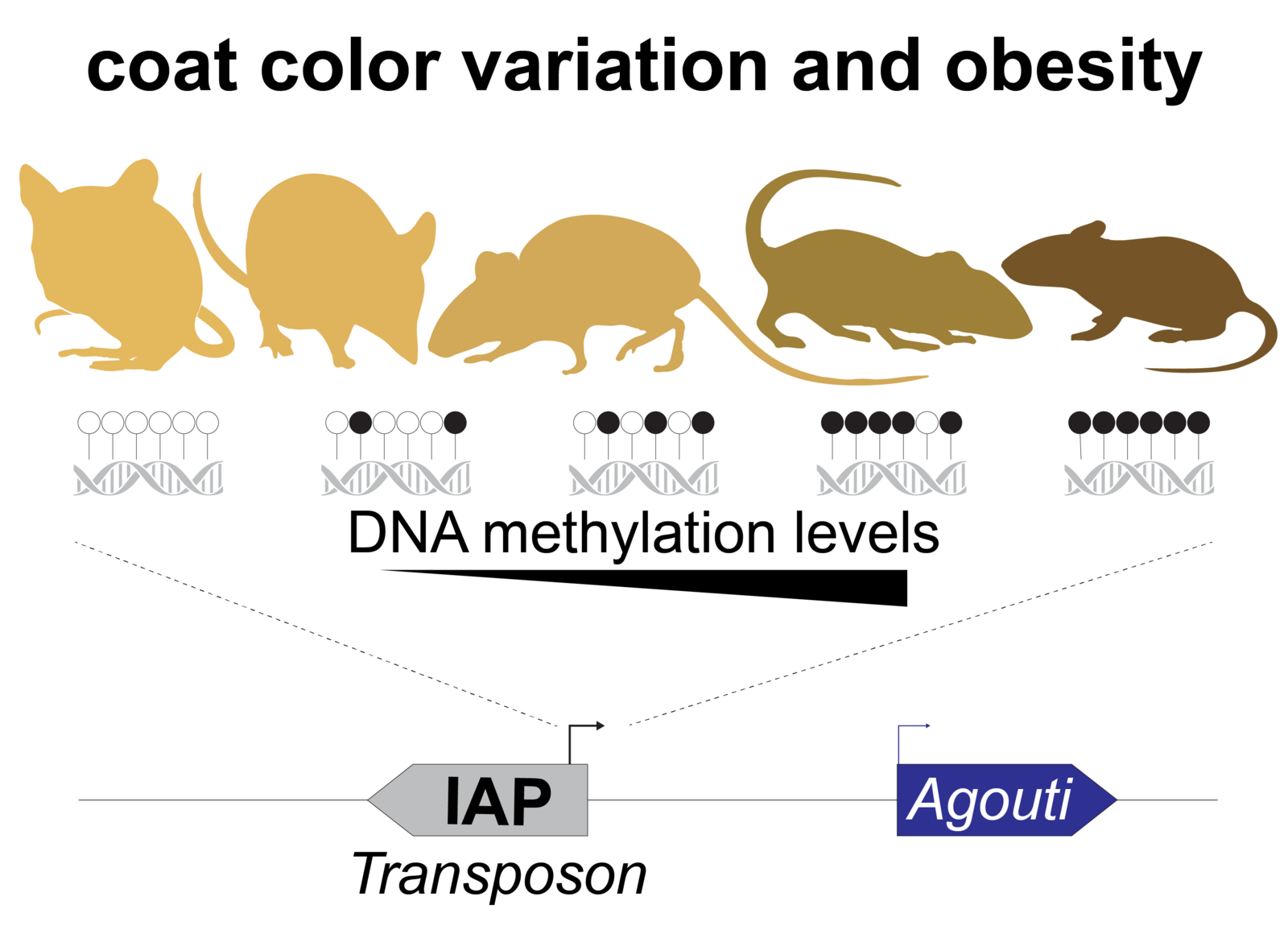
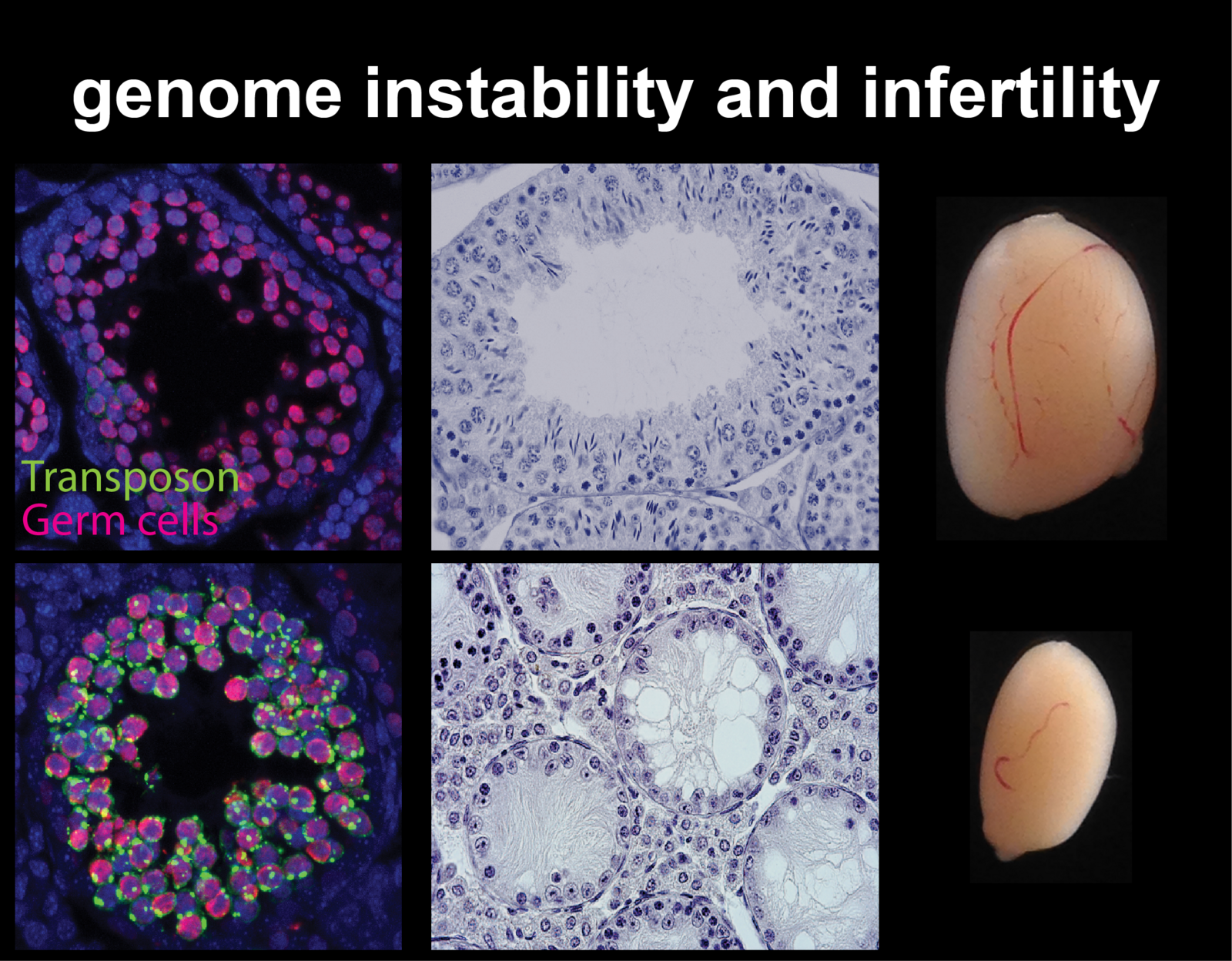
Transposable elements, or transposons, are abundant genomic repeats discovered initially in maize by Barbara McClintock, work for which she was awarded a Nobel prize in 1983. She recognised them in the early 1950s through their link with genome instability and their ability to control and disrupt genes with phenotypic consequences. The extension of modern evolutionary synthesis to molecular biology brought with it its gene-centric view of evolution and quickly identified transposons as a classic example of selfish, parasitic DNA. They are said to be agents of intragenomic conflict because they drive their heritability, increasing their frequency in a population regardless of any benefit to the host genome. On the contrary, many examples of their mutagenic activity with harmful consequences to genome functions have been documented. The evolution of mechanisms for transposon control and the co-evolution of ways transposons subsequently evade them is effectively an arms-race. This race has been shaping how our genome functions for billions of years and is reflected in nearly every aspect of our biology.
Transposons, epigenetics & genome instability
Cytosine methylation is an epigenetic DNA modification correlated with transcriptional silencing. Mammalian genomes make widespread use of DNA methylation for the long-term repression of transposons. Altered DNA methylation levels at transposons can lead to various degrees of reactivation, with consequences ranging from altered gene control, leading to phenotypic variation within an isogenic population (e.g. Agoutivy epiallele), to catastrophic genomic instability and cell death, e.g. infertile mutants of germline DNA methyltransferases. Transposon sequences make up to 50% or more of our genomic DNA, and genetic and epigenetic variation at these sequences have an enormous potential to influence patterns of inheritance and how our genome functions both in normal development and in disease.
What we love to do
Our lab primarily studies how transposon biology impacts our genomes. In particular, the genome of germ cells and pluripotent stem cells. We are interested in uncovering new mechanisms by which genomes attempt to control transposons and how transposons escape them. We are also interested in dissecting how transposons themselves and the mechanisms originally involved in their regulation can be brought in to play important roles in development and disease. Using the mouse and cell culture as models, we combine biochemistry, genetics, genomics and developmental analysis to study the genetic conflict with transposons, uncovering fundamental aspects of germline biology and genome regulation and instability.
Our Projects
Uncovering the regulatory arms-race between transposons & the germline
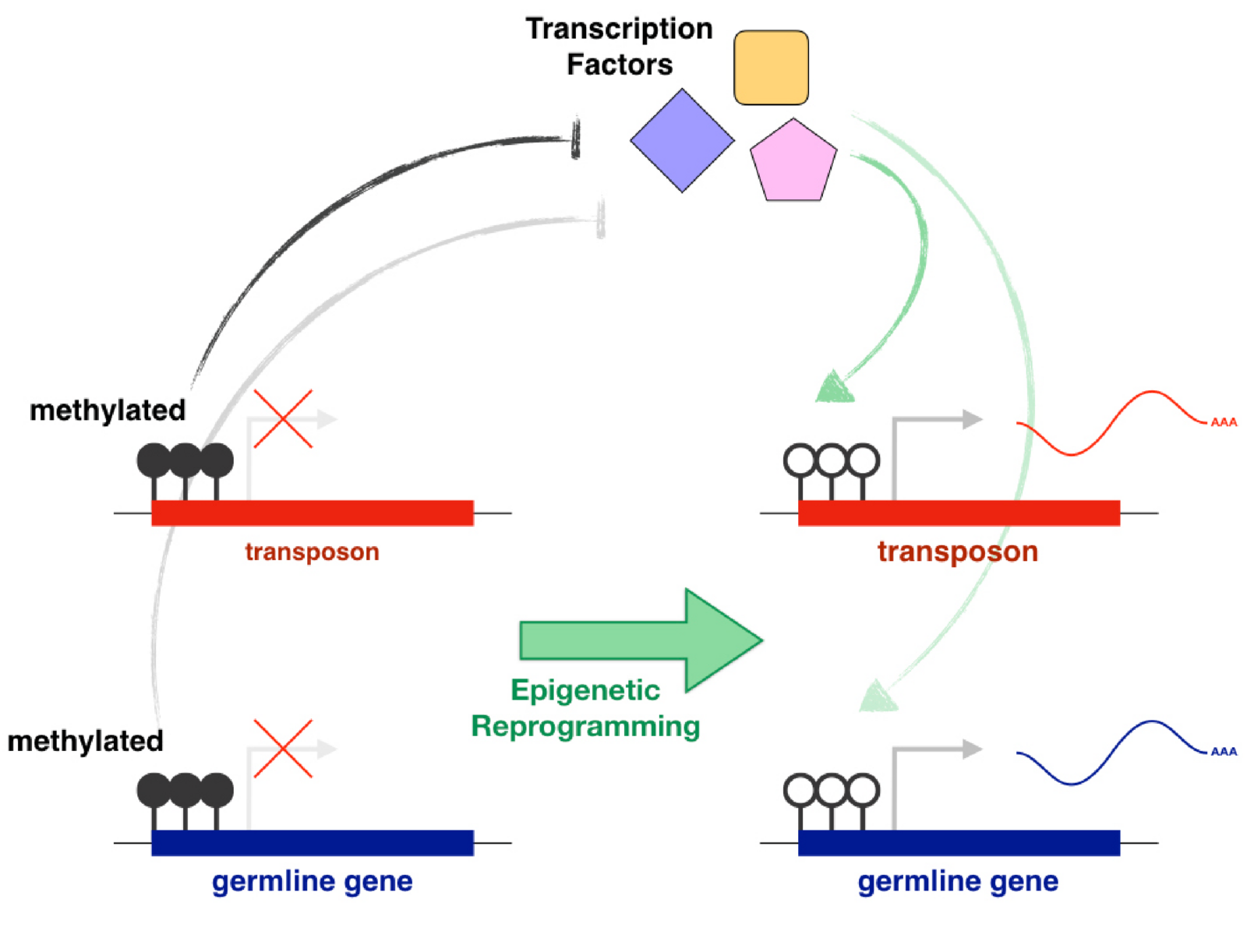
The main examples of direct gene regulation by DNA methylation during development are the silencing of germline genes in somatic cells and the silencing of transposons. Germline DNA methylation reprogramming allows the expression of germline genes and gametogenesis. However, it also releases epigenetic repression from transposons, putting germline integrity at risk. Transposons drive their heritability by being active in the germline and evolution must favour copies that are able to use germline transcriptional activators. In light of this, the effective role of promoter DNA methylation in the repression of both germline genes and transposons is likely not a coincidence, but rather a direct consequence of intragenomic conflict. Our goal is to identify and characterise the transcriptional activators of transposons and germline genes. We also want to understand how their activity is affected by the epigenetic state of their targets and how this is regulated across development. Finally, we will use comparative genomics and in vitro functional assays to explore how a strong selective pressure to avoid binding to transposons affects the evolution of germline transcriptional networks that are essential for reproduction (e.g. meiosis).
The epigenetic regulation of transposons in the germline
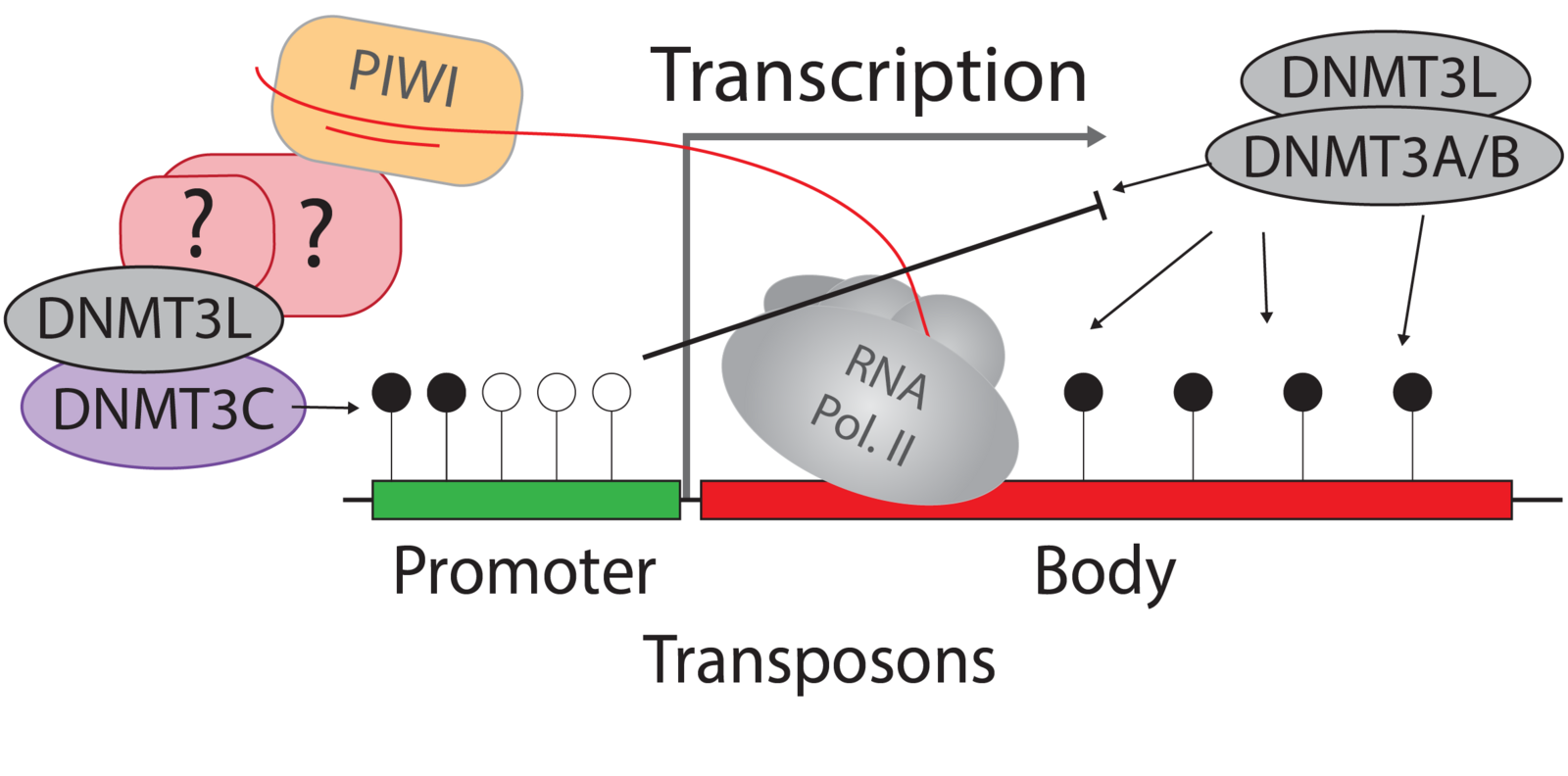
Germ cells ‘record’ which transposon copies are active by processing their mRNA into small RNAs. These are called Piwi-interacting RNAs (piRNAs) because they require and associate with PIWI argonaute proteins. PIWI proteins constitute the core of genome defence against transposons. They connect the biogenesis of piRNAs with the effector mechanism that places DNA methylation at active transposon copies and stably silences them. The biology of piRNAs is an intense field of study. However, the effector mechanism connecting PIWI proteins with de novo methylation has remained elusive. The recently discovered DNA methyltransferase enzyme DNMT3C specifically methylates the promoters of transposons in germ cells, and Dnmt3C mutants are infertile due to massive transposon reactivation in germ cells. Their phenotypic and methylation defects entirely match those found following mutations of PIWI proteins, suggesting DNMT3C as the most downstream component of this pathway known to date. We combine mouse genome editing tools and current, innovative biochemistry methods to identify the proteins involved and the mechanism allowing piRNA-guided DNA methylation at promoters.
Spoils of war: genome defence beyond transposon regulation in the germline

One important consequence of the fast-paced evolutionary arms-race between the genome and transposons is the creation of fertile ground for the evolution of biological novelty. Small RNAs and epigenetic modifications have been described in many eukaryotes as agents contributing towards the inter-generational inheritance of genome regulation. Despite reports of non-DNA sequence-based inheritance in mammals, a mechanistic framework for studying how small RNAs are used to convert information into regulation across generations is notably absent. In addition, with the exception of genomic imprinting and variegated transposon DNA methylation, we currently don’t really know if this can happen naturally in mammals and what are the extents of its consequences in terms of genome regulation and health and disease in the following generations. To explore these questions, we develop functional genetics tools in order to manipulate small RNAs along with readers and writers of epigenetic information. We carry this out in early embryos and combine it with fine molecular and developmental analysis of the consequences. We aim to identify new mechanisms in this system and put them to the test within a consistent framework of intergenerational transmission of instructional information for genome regulation.
Recruiting
Our lab is growing to be a multi-cultural, open, dynamic environment. We always welcome the chance to discuss new ideas and their implementation in the framework of our research. We value motivation and passion for science and the topics of our research over a perfect CV, and we strive to provide consistent mentoring and career development to our members. Learn more about positions and projects by sending us an email here.